信息中心



在过渡金属催化领域,通过计算进行催化剂的高通量虚拟筛选是实现催化剂理性设计的重要途径。近年来,基于配体描述符的机器学习策略得到了广泛发展,其核心在于通过拟合实验数据来近似预测催化性能;另一种更具普适性的路径则是基于第一性原理,通过直接计算反应能量图谱来评估催化剂表现。然而,要实现基于第一性原理的反应路径全自动虚拟筛选,目前在学术界仍面临三个关键瓶颈:一是过渡金属配合物、反应中间体和过渡态(TS)的三维结构数据极度匮乏,传统的理论数据集规模有限且高度依赖人工搭建与收集;二是缺乏能够同时兼容稳定中间体和过渡态、且能拓展至百原子以上大体系的统一三维结构生成框架;三是中间体和过渡态自由能的精确评估成本高昂,传统DFT计算效率不足。
近日,针对这些瓶颈,h小说 刘智攀教授团队在《美国化学会志》(J. Am. Chem. Soc. )上发表了研究论文(//pubs.acs.org/doi/10.1021/jacs.6c03190)。该研究提出了一种耦合势能面探索的自学习扩散模型框架(SL-DM-PES),系统性地为上述有机催化预测瓶颈提供了高效的解决方案。该论文是继近期课题组在拓展人工智能原子模拟在无定形结构预测方向(J. Am. Chem. Soc. 2026, 148, 23088, //pubs.acs.org/doi/10.1021/jacs.6c05023?ref=PDF) 取得的又一重要进展。
针对结构数据匮乏的问题,该框架开发了一种闭环的自学习迭代机制(SL-DGM-PES)。该机制巧妙地将生成式AI和势能面方法相结合,从二维分子图出发,通过自动化的结构生成、势能面优化和知识库动态更新,自主构建了包含 105,370个金属配合物的 PdP8数据集,实现了过渡金属催化反应空间的高效采样与数据积累。
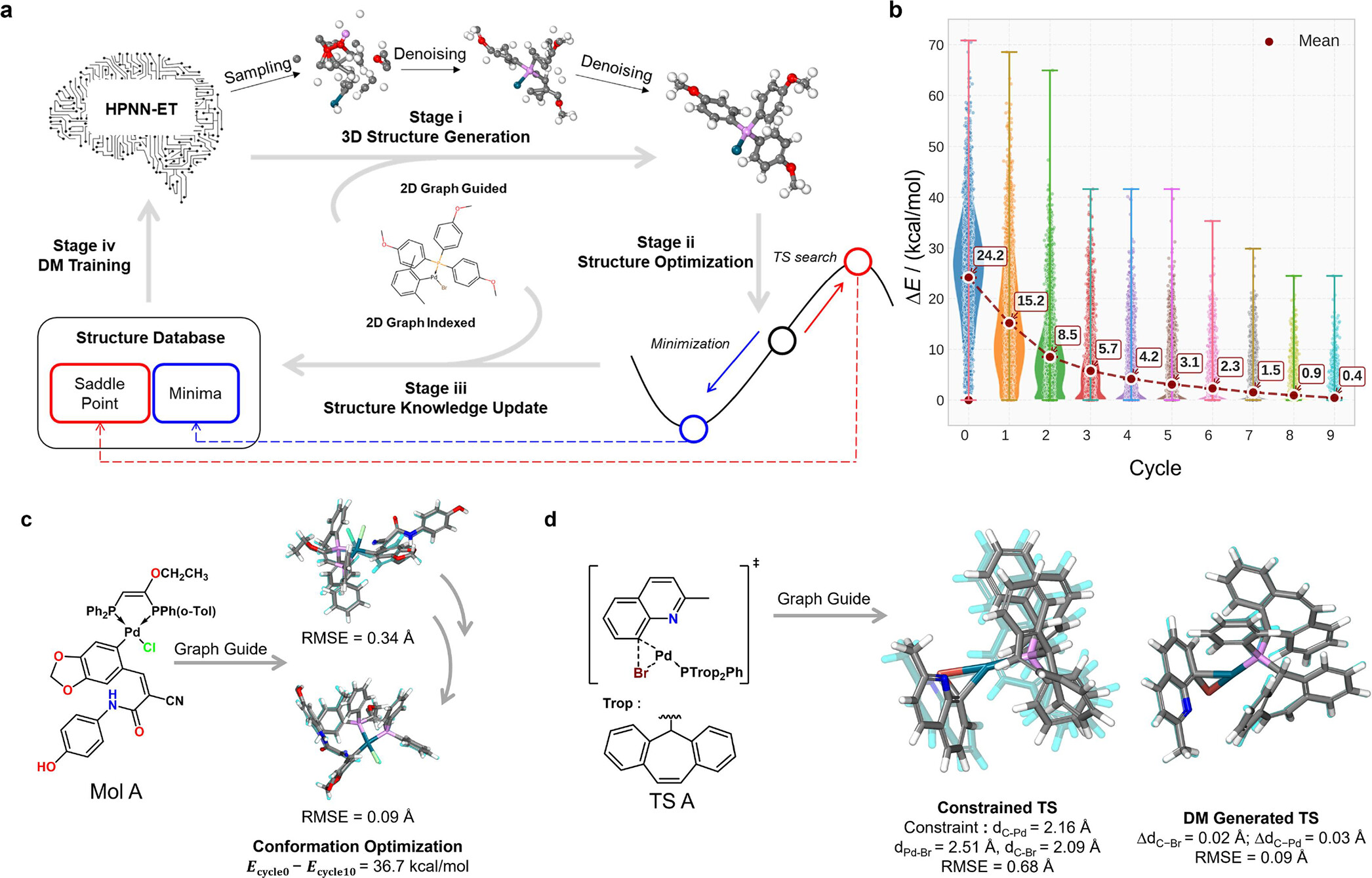
图1 使用SL-DM-PES框架自动探索未知化学空间,生成高质量结构数据集
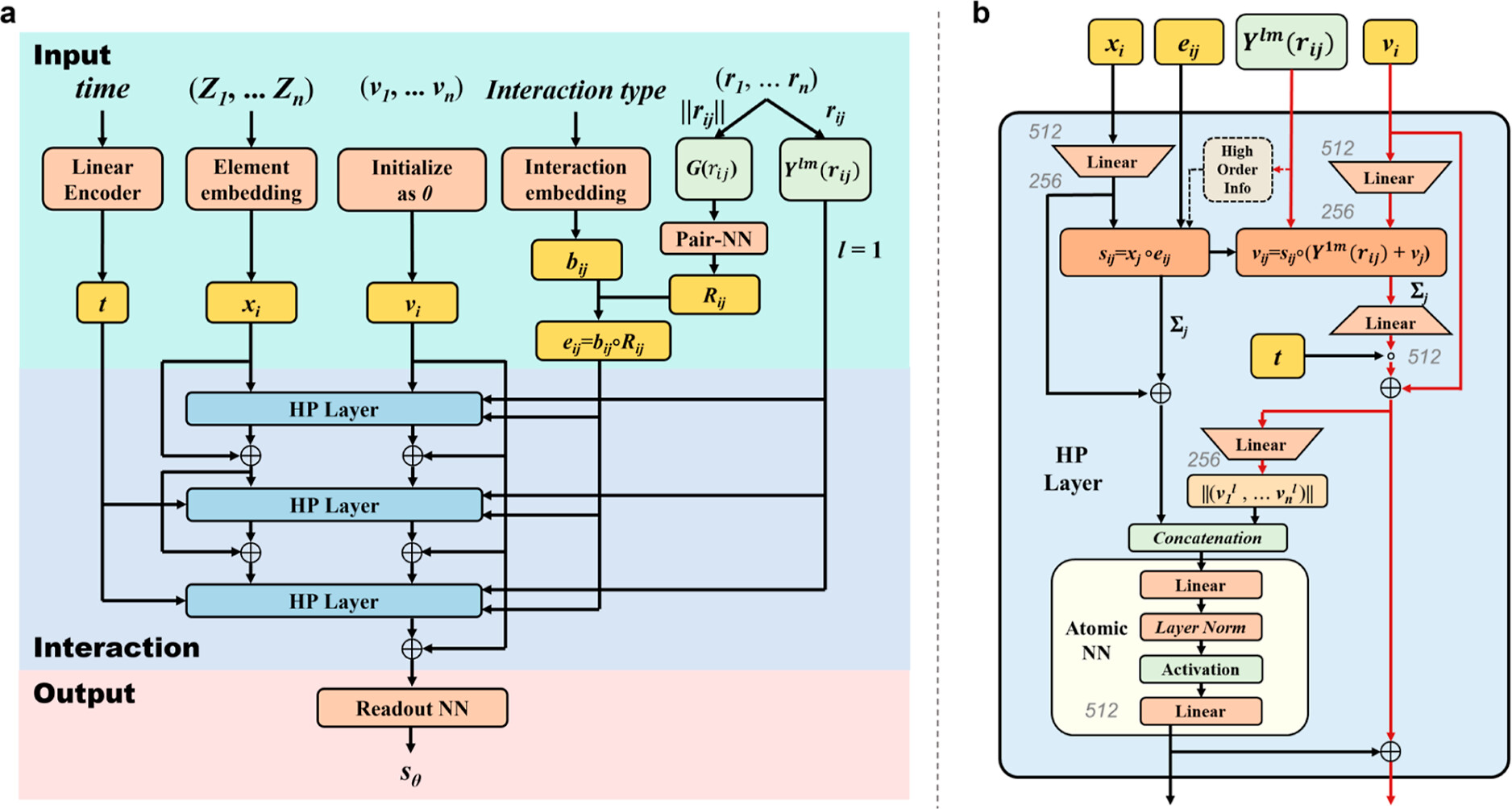
针对大体系和过渡态统一生成的难题,研究团队设计了高阶对收缩等变时空消息传递神经网络(HPNN-ET)作为扩散模型的核心。该模型在结构生成精度与计算速度上均表现出极其优异的性能:在保持原子级超高精度(中位数均方根偏差 RMSE ≤ 0.062Å)的同时,实现了极高的生成效率(单结构生成时间最快仅需 0.35秒)。该网络采用统一的信息融合策略,将二维拓扑与三维几何信息同步处理,并通过在化学键类型指标中显式编码成键和断键信息,将过渡态与稳定分子纳入统一表征。同时,通过在消息传递中引入维度缩减与恢复机制,显著降低了计算复杂度,成功实现了对超大配位体系的高效外推。
针对能量评估与过渡态定位的成本问题,该框架将扩散模型与团队前期发展的通用全局神经网络势(GG-NN)深度耦合,利用GG-NN对扩散模型生成的初始结构进行快速结构优化,并结合Constrained Broyden–Dimer (CBD)算法进行过渡态的精确本地定位与验证,在保持DFT级结构精度的同时,极大地缩短了计算时间与降低了经济成本。
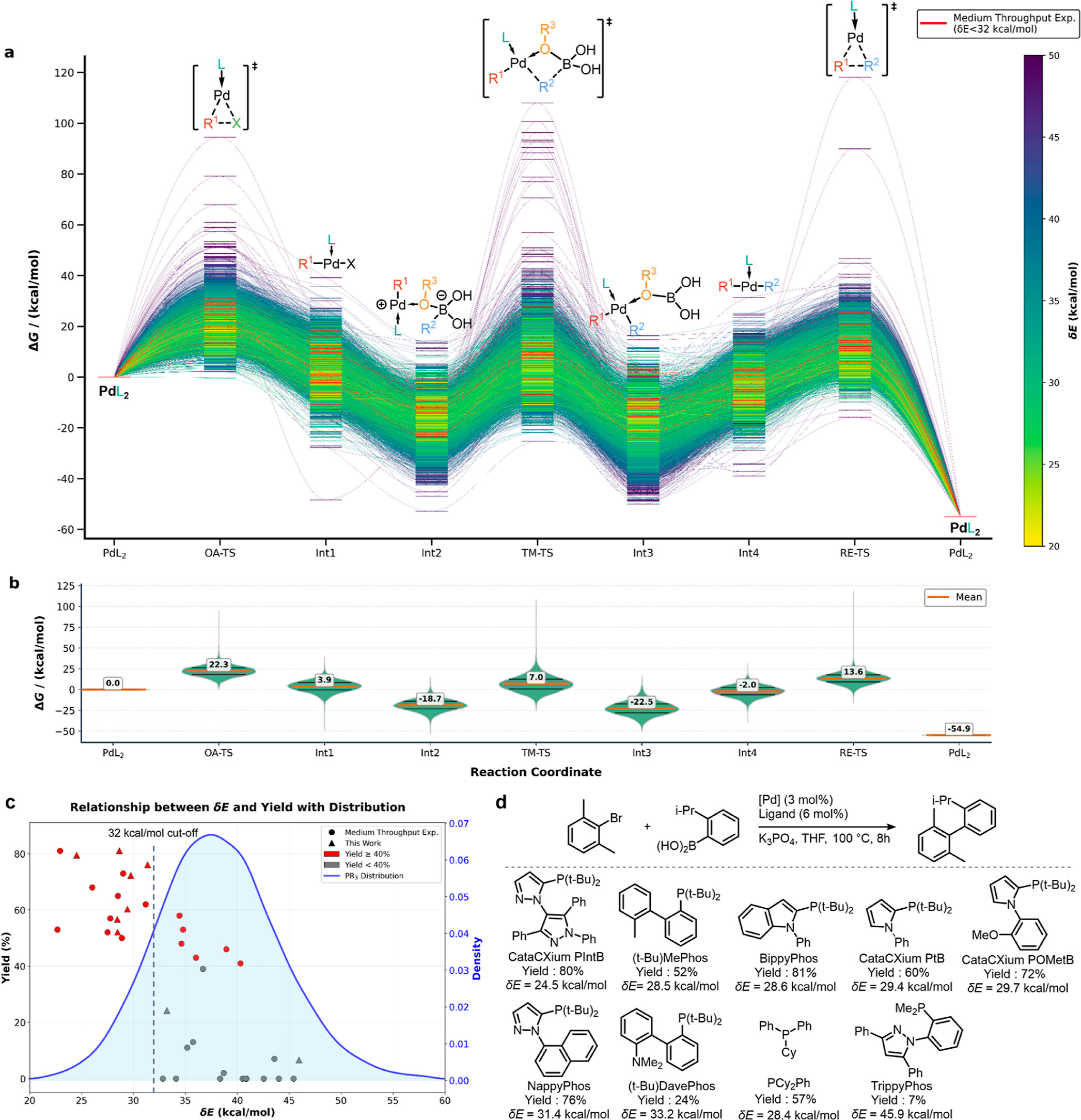
作为应用实例,研究团队将SL-DM-PES框架应用于经典的钯催化Suzuki-Miyaura交叉偶联反应。在仅消耗286个GPU小时(RTX 4090)、总体花费约80美元(折合每个催化剂约0.01美元)的条件下,全自动构建了6883种不同膦配体催化剂的完整反应自由能图谱。基于大规模能量数据,研究团队提出了“催化活性温和带”(CAT-Band)概念,阐明了优秀催化剂需在中间体稳定化与过渡态能垒之间取得热力学与动力学平衡的物理本质。通过快速二分类筛选,团队预测并实验证实了CataCXium PIntB、BippyPhos等高活性商业化配体(实验产率达72%~81%),计算趋势与实验高度吻合。
目前,该框架的核心扩散模型已集成至LASPAI人工智能原子模拟平台(//www.laspai.com/)并开放线上操作。该工作通过解决数据丰度、模型可扩展性与能量评估效率三大瓶颈,为基于第一性原理的催化剂全自动、机制知情的高通量筛选提供了可行路径。
图3 基于Pd催化的Suzuki-Miyaura交叉偶联反应,通过全自动反应路径扫描与动力学能垒评估实现配体高通量筛选